Due to the possibility of studying cell states and effect of master regulators and possibility of cell response prediction, different research groups have started producing large dataset of single cell open chromatin profiles. The increase in dataset of single cell open chromatin profiles has created a need for search engine which can provide match with existing single cell expression and epigenome datasets. However unlike expression profiles, where genomic loci (as genes) are fixed, single cell epigenome profiles from different groups do not have signal from same locations. Hence there is need to have specialised search engine which can bring uniformity in analysing epigenome signals in non-similar genomic loci. With ScEpiSearch users can query with single-cell open-chromatin profiles with their own peak list, to retrieve the top matches from a large database of the single cell expression and epigenome profiles with relevant meta-information.
Note : Cross-batch and Cross-species embedding is available in stand-alone version.
Steps of use :
ScEpiSearch has a user-friendly interface. This page gives a step by step guide on how to use the search engine. Please go through the steps mentioned below to understand the procedure to be followed in order to find similar items to your query samples
Step-1
Select the dataset (human / mouse) you want to search against.
Step-2
Fill in the Email ID in the form where you wish to receive the result page URL (Optional).
Step-3
Enter the number of nearest neighbours you wish to find for your query.
Step-4
Upload the query file. For query file guidelines, see 'Input File Preparation' Section.
Hit the SUBMIT Button.
Step-5
Once your query is completely processed, the job status page is displayed. Navigate to the result page by clicking on view results on job status page.You will see a page shown as shown below.
Step-6
Once your query is completely processed, you can click on result button to view results.
Input file description
File Format
ScEpiSearch accepts two file inputs from user for a query i.e Count Epigenome file containing tag counts for various cells to be queried and peak chromosome locations common for all cells in count file.
Count file should be comma separated file (.txt) files in zipped (.txt.zip , .txt.gz) formats. The query file should not contain any header or row indices.
Chromosome file should be tab separated bed file in zipped format (.bed.zio, .bed.gz)
File Description
User should provide two files, first file should contain tag-counts on genomic locations (peaks) and second file has location of peaks.
Interpretation of output
For single cell open chromatin profile query, ScEpiSearch provides two types result of match. First type of results show top matching cells using single cell expression profiles in reference dataset. The second result show matching cell using their opne-chromatin profile.
Clickable World Cloud :
The word cloud gives a complete overview of all matching phenotypes occuring in both epigenome and expression matches for all queries.
Top 50 Enriched Genes :
On clicking the word cloud image, there appears a pop up window of top 50 enriched genes and its MGI equivalent for query (default is for query 1). When clicking on the details of gene, user gets description of celltype iformation of which the gene is a marker. Using the drop down user can search for any query.
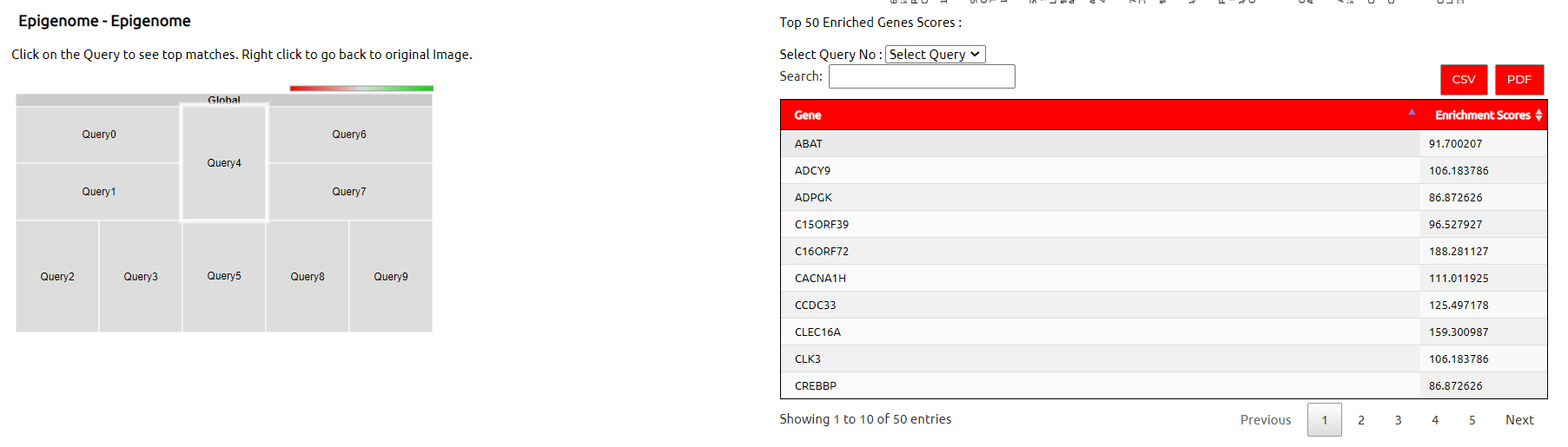
Interactive (Clickable) SOM Plot :
SOM map of queries is made based on projection on epigenome reference cells. User can click on query points to view further details.
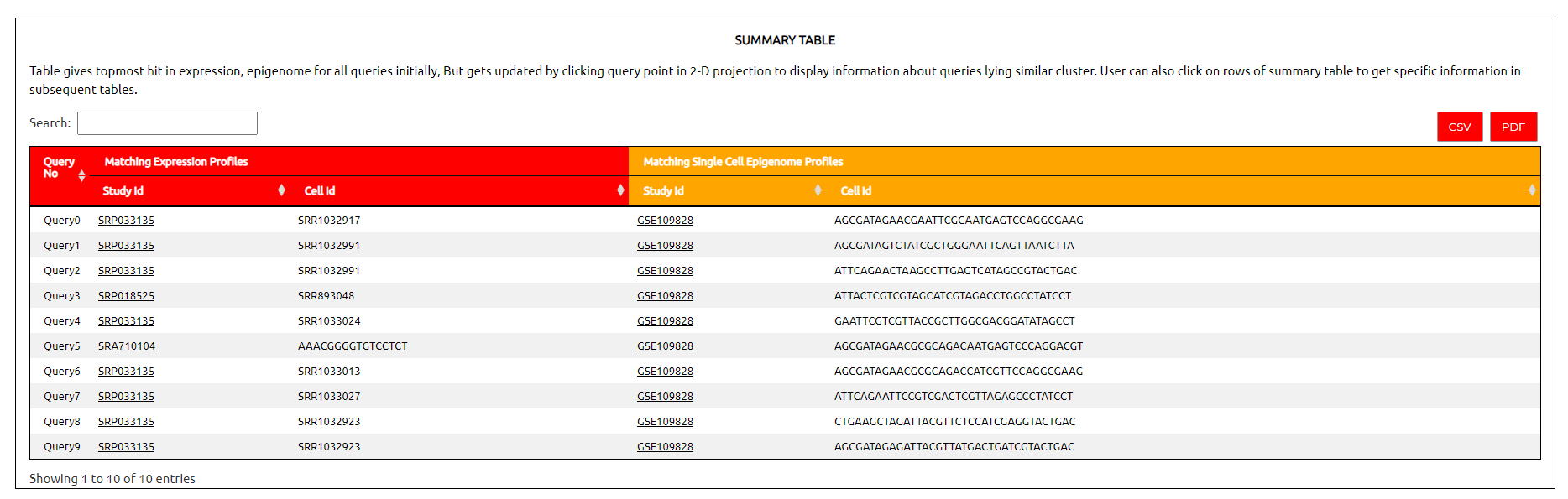
Summary Table :
The summary table shows topmost match in epigenome and expression match for every query. Rows of Summary table are clickable.
Interactive Summary :
This is a google tree-map inspired plugin which gives an interactive visualiztion of viewing frequent repeating discription. While left clicking on the block of query one can see the highest occuring discription in the result for the given query. Left click to go back to original image.
Gene Frequency Bar Plot :
The frequency plot shows frequency of genes in all queries.
Enrichment Scores for top Enriched Genes of Query :
This table displays enrichment scores for top enriched genes.
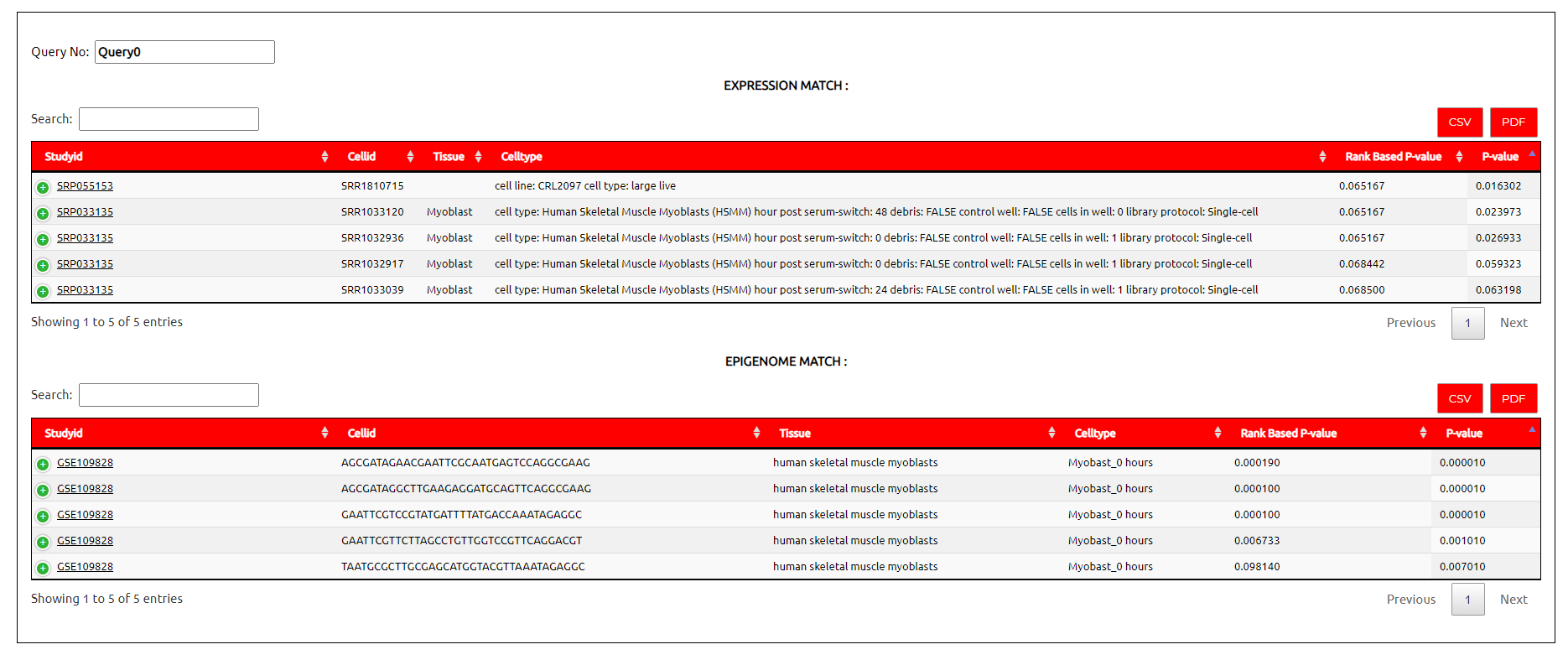
For match with single cell expression profile it provides :
P-Value :
For each matching cell in reference it provides the statistical significance of match in terms of P-value. Here P-value shows chances of occurrence of median normalised expression of proximal genes in that cell type was purely by chance or not.
Rank based adjusted P-Value :
Once P-value is calculated, the rank of a reference cell for a query cell is used to calculate new rank based P-value.
Study ID :
For each cell in the result, this column gives the study or experiment from which it is was curated. The text in this column is linked to its SRA study page.
Description :
This column provides information about the matching cells
For match with single cell epigenome profile it provides
P-Value :
For each matching cell in reference it provides the statistical significance of match in terms of P-value. Here P-value shows chances of occurrence of correlation in that cell type was purely by chance or not.
Rank based Adjusted P-Value :
Once P-value is calculated, the rank of a reference cell for a query cell is used to calculate new rank based P-value.
Study ID :
For each cell in the result, this column gives the study or experiment from which it is was curated. The text in this column is linked to its SRA study page.
Description :
This column provides information about the matching cells
Heatmap
The heat-map is generated by taking the correlation values of the top hits for each query cell. This union of top hits is listed on the y-axis, and the input cells are listed on the x axis to form a grid. Each element in the grid shows the intensity of correlation values for the predicted hit for that input query cell.